 B4.1: Small lock-and-key elements to track and manipulate receptors in time and space
B4.1: Small lock-and-key elements to track and manipulate receptors in time and space
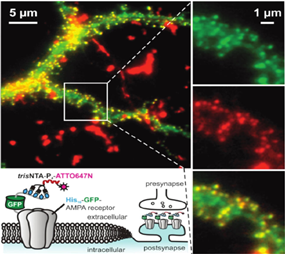 The PhD student will explore wavelength-addressable optochemical tools for the spatiotemporal labeling and assembly of proteins. Novel small lock-and-key elements will be developed in order to site-specifically label, track and manipulate receptor proteins in time and space. The photophysical properties (e.g. uncaging quantum yield, two-photon cross-section) of the light-sensitive moieties will be analyzed and optimized in close collaboration with A2.1. Further performance tests of the newly developed PA-trisNTAs will be undertaken at functionalized interfaces (surface plasmon resonance, TIRFM, etc.). By strategic positioning of black-hole quenchers, photo-cleavage will cause fluorescence dequenching and allow high affinity labeling upon illumination for super-resolution microscopy (in collaboration with B5.1).
The PhD student will explore wavelength-addressable optochemical tools for the spatiotemporal labeling and assembly of proteins. Novel small lock-and-key elements will be developed in order to site-specifically label, track and manipulate receptor proteins in time and space. The photophysical properties (e.g. uncaging quantum yield, two-photon cross-section) of the light-sensitive moieties will be analyzed and optimized in close collaboration with A2.1. Further performance tests of the newly developed PA-trisNTAs will be undertaken at functionalized interfaces (surface plasmon resonance, TIRFM, etc.). By strategic positioning of black-hole quenchers, photo-cleavage will cause fluorescence dequenching and allow high affinity labeling upon illumination for super-resolution microscopy (in collaboration with B5.1).
Suggested literature:
- "Nanomolar interaction and in-situ assembly of proteins in four dimensions controlled by light", N. Laboria, R. Wieneke, R. Tampé, Angew. Chem. Int. Ed. 2013, 52, 848-853.
- "Quantum yield optimized fluorophores for site-specific labeling and super-resolution imaging", C. Grunwald, K. Schulze, G. Giannone, L. Cognet, B. Lounis, D. Choquet, R. Tampé, J. Am. Chem. Soc. 2011, 133, 8090-8093.


B4.2: In-situ assembly of receptors triggered by light
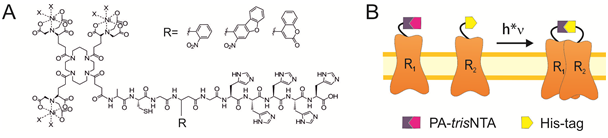
This project aims at introducing the newly designed PA-trisNTAs into cell surface receptors by bioorthogonal labeling. Based on the self-inactivation, the PA-trisNTA and His-tagged receptors will interact only upon light activation, which can be performed in dual or triple color mode by LSM or TIRF microscopy. By the generation of protein binding regions upon illumination, the process of iterative protein binding and sequential clustering of membrane receptors will be examined. The project will take advantage of a range of receptors fused to a fluorescent reporter protein and advanced microscopy in collaboration with B5.1.
The PhD student will be able to control receptor clustering in vitro and later in vivo. The lateral organization of membrane receptors will be realized in giant unilamellar vesicles (GUV), solid supported membranes, as well as the plasma membrane of living cells. The receptor mobility before and after photo-activation will be analyzed with fluorescence correlation spectroscopy (FCS), fluorescence recovery after photobleaching (FRAP), and single-particle tracking in combination with photo-switchable fluorophores (in collaboration with B5.1).
Suggested literature:
- "Integrins function as homotypic stable adhesion units transiently trapped in adhesion sites", O. Rossier, V. Octeau, J. B. Sibarita, C. Leduc, B. Tessier, D. Nair, V. Gatterdam, O. Destaing, C. Albiges-Rizo, R. Tampé, L. Cognet, D. Choquet, B. Lounis, G. Giannone, Nat. Cell Biol. 2012, 14, 1057-1067.
- "In situ assembly of macromolecular complexes trigged by light", C. Grunwald, K. Schulze, A. Reichel, V. U. Weiss, J. Piehler, K. H. Wiesmüller, R. Tampé, Proc. Natl. Acad. Sci. USA 2010, 107, 6146-6151.

B4.3: 3D organization and manipulation of proteins by light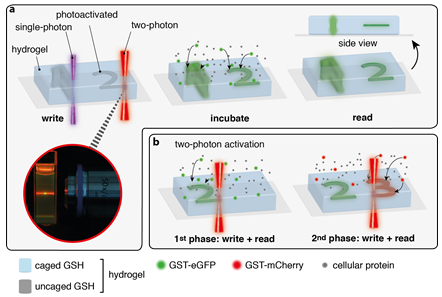
This project will focus on the in-situ assembly of biomolecules in 3D scaffolds. Laser scanning lithography combined with light-triggered protein assembly offers a powerful tool to organize protein gradients and clustering for a broad range of applications. The PhD student will employ the site-specific and spatiotemporal organization of proteins as well as the sequential protein clustering in 3D scaffolds. For elaborated applications, the photophysical properties, like wavelength selective-activation and two-photon accessibility will be systematically optimized (with A2.2 and A3.2). The process of iterative binding and sequential protein clustering of proteins, including the fabrication of lateral protein gradients will then be examined. Mask-patterned illumination and laser lithography will be employed to write in-situ regions and protein gradients.
Figure: In-situ protein assembly by optochemical tools. A) Comparison of one and two photon activation in hydrogels by light (a) and three dimensional protein assembly by two photon laser-scanning microscopy (b).
Suggested literature:
- Gatterdam V, Stoess T, Menge CM, Heckel A, Tampé R (2012) Caged glutathione – triggering protein interaction by light. Angew Chem Int Ed 51, 3726-9.
- Roizard S, Danelon C, Hassaine G, Piquet J, Schulze K, Hovius R, Tampé R, Vogel, H (2011) Activation of G protein coupled receptors in native plasma membrane supported on beads. J Am Chem Soc 133, 16868-74.

B4.4: Opto-control of receptor clustering and synthetic photo-conditional proteins
In the first period of CLiC, Alina Klein (née Kollmannsperger) successfully delivered small lock-and-key elements to the cytosol of living cells for the spatiotemporal tracking of proteins. Cargo delivery was efficiently facilitated by microfluidic cell squeezing and enabled the transfer of a plethora of lock-and-key elements as well as light-addressable modules. This allowed to precisely control the site-specific labeling, tracking and manipulation of receptor proteins at single-cell level by light and culminated in the live-cell super resolution of the nuclear lamina by high affinity multivalent chelator heads. In close collaboration with the Heilemann lab, the live-cell fluorescence microscopy was performed and the photophysical properties as well as the fluorogenic behavior of the developed light-sensitive moieties were analyzed in contact with A2.1.
In parallel, Karl Gatterdam aimed at developing new photo-conditional, ultrasmall lock-and-key pairs for the organization of receptor proteins by light. He engineered chemical inducible dimerizers based on peptide as well as DNA platforms and was further able to generate multivalent chelator heads, which exhibit subnanomolar to picomolar affinities (super binder). Chemical induced dimerization was analyzed in contact with Tobias Lieblein (B6).
On the basis of these achievements, we will explore the newly established lock-and-key elements for the rapid fluorescence labeling of receptor proteins in living cells. The PhD student will work on the light-mediated, traceless modification of receptors as well as the site-directed conjugation of single-stranded DNA/PNA (finally caged strands) to cell surface receptors. The analysis of the traceless receptor modification will be performed
by live-cell and high-end fluorescence microscopy. To promote the light driven dimerization/oligomerization of receptors, real-time confocal laser scanning microscopy will be performed and the start of protein-protein interaction will be tested at different time points by illumination in live, intact cells. In addition, the extension of the UV-controlled assembly of proteins at functionalized interfaces (PEGylated SiO2, PLL-PEG surfaces) as well as three-dimensional scaffolds (e.g. DNA origami, hydrogels) will be explored to control protein assembly and ultimately receptor clustering at the molecular level. To gain even higher spatiotemporal resolution for site-specific protein trapping, optochemical tools will be designed, which can be photo-activated even by two-photon processes (in contact with A2.6).

B4.5: Photo-triggered reorganization of the PLC and related MHC-I chaperone complex
Constant contact to pathogens, such as bacteria, fungi, and viruses, has led to the development of the sophisticated self-defense system of adaptive immunity. Infected cells expose a signal to initiate their recognition and destruction by responsible immune system cells. The constant turnover of the cellular proteome results in antigenic peptides, which are loaded onto major histocompatibility complex class I (MHC-I) molecules. The process of loading is accomplished by the peptide-loading complex (PLC). At the heart of this dynamic multiprotein machinery is the ABC transporter associated with antigen processing that mediates translocation of cytosolic peptides into the endoplasmic reticulum (ER). Despite of MHC-I, the multi-subunit complex comprises several ER chaperone and quality control proteins responsible for stability and peptide editing. After rounds of processing, stable peptide-loaded MHC-I complexes are released for their journey to the cell surface, where they are recognized by cytotoxic T-cells. Recently, the structure of the PLC and other MHC-I/chaperone were determined by cryogenic electron microscopy highlighting its individual components.1,2 However, the spatiotemporal structure-to-function interplay between MHC I and various chaperones remains elusive. Therefore, Ines Katharina Müller aims to utilize a modular photo-conditional approach (in collaboration with B4.6) to trigger the assembly of MHC-I chaperone and peptide loading complexes. Finally, this studies will provide fresh insights into mechanistic principles of adaptive immunity using light as orthogonal trigger.
[1] Blees A, Januliene D, Hofmann T, Koller N, Schmidt C, Trowitzsch S, Moeller A, Tampé R (2017) Structure of the human MHC-I peptide-loading complex. Nature 551, 525-8. (Cover Story and News & View in Nature; Dispatch in Current Biology; Highlight in F1000)
[2] Thomas C, Tampé R (2017) Structure of the TAPBPR-MHC I complex defines the mechanism of peptide loading and editing.

B4.6: Photo-conditional immune suppressors
In adaptive immunity, major histocompatibility class I (MHC I) molecules are loaded in the ER lumen with proteasomal degradation products for antigen presentation on cell surfaces. As a key component of the peptide loading complex (PLC), the transporter associated with antigen processing (TAP) plays a prime role by supplying antigens for loading of MHC I.
Building on our recent cryo-EM and X-ray structures of the human PLC and related MHC I chaperone complexes, we will explore photo-conditional viral immune evasins to control the function and conformation of TAP. Major aims are the assembly and disassembly of the PLC in its active and virally arrested state as well as the molecular architecture and dynamic reorganization of the PLC from its arrested to its active state. To spatiotemporally control activity of the viral immune evasin and thus PLC-mediated antigen processing, Christian Winter will develop photo-conditional immune suppressors. He aims to design semi-synthetic and fully synthetic viral peptides. We plan to introduce up to two novel photo-cleavable red-shifted β-amino acids (in collaboration with Heike Krüger, A7.1, Wieneke group) for photo-activation.
Viral activity and TAP-specific antigen translocation will be analyzed by the ultrasensitive single-cell peptide translocation assays in vitro and subsequently in vivo (live-cell microscopy, real-time CLSM). Ultimately, the control of protein-protein interaction, localization and antigen translocation in living cells will be analyzed with fluorescence correlation spectroscopy (FCS), fluorescence recovery after photobleaching (FRAP), and single-particle tracking and, whenever possible, by super-resolution microscopy.
Suggested literature:
C. Thomas, R. Tampé, Science 2017, 358, 1060-1064. “Structure of the TAPBPR–MHC I Complex Defines the Mechanism of Peptide Loading and Editing”
A. Blees, D. Januliene, T. Hofmann, N. Koller, C. Schmidt, S. Trowitzsch, A. Moeller, R. Tampé, Nature 2017, 551, 525-528